














Neutralizing Antibodies in the Fight against COVID-19
 Publication Date:2021-02-16
Publication Date:2021-02-16 Page Views:7018
Page Views:7018COVID-19 neutralizing antibodies (NAbs) have been one of the big stories of the coronavirus pandemic. Produced by immune system B cells, NAbs stop infections by incapacitating the invading pathogen. From the moment their discovery was announced in May 2020, coronavirus NAbs were touted as possible treatments and diagnostic targets.
SARS-CoV-2 NAbs usually form around 1-2 weeks after infection. Serologic tests thus expand the testing time window beyond the acute infection phase, but they cannot diagnose acute infections. The assays also help track infection chains and answer clinical, virological, and epidemiological questions, including how many infected individuals remain asymptomatic, and whether a robust antibody response has occurred.
Almost all coronavirus NAb assays carry the research use-only (RUO) designation. RUO does not imply any level of adherence to regulatory or quality directives, and products designated as such carry no warrantee except for their contents. Documentation, validation, or certifications of purity or quality are provided for product differentiation, not to satisfy regulations.
By contrast in vitro diagnostics (IVDs) are approved, highly regulated medical devices whose development follows ISO 13485 guidance as well as jurisdictional regulations like CE marking and the newer European In Vitro Diagnostic Regulation.
An exercise in risk management
Patient variability always makes the journey from RUO to IVD challenging, and coronavirus NAbs are no exception. "The concentration of NAbs in blood varies greatly from person to person," says Kai Fechner, Director of International Sales and Marketing for EUROIMMUN, a PerkinElmer company.
A wide range of NAb titers means that correlations between antibody levels and disease or immunity status becomes murkier, and in the world of health regulation murkiness equals risk.
"Regulators of IVDs are concerned mainly with risk, and that the device does what it promises, nothing more and nothing less," says Marcus Manocha, Custom Services Manager at Absolute Antibody. "The performance data required depends on the complexity and type of test, which include clinical data supporting limits of detection, reproducibility, and performance in the presence of interfering substances. Commercializing an IVD is not trivial. It requires significant capital plus a network of teams working toward a common goal. At its core IVD approval is a task in risk management."
As with therapeutics based on coronavirus NAbs, regulators have made exceptions to normal approval protocols for COVID-19 serologic (antibody) diagnostics, which the Agency approves through Emergency Use Authorizations (EUAs).
FDA issues EUAs as needs arise, bypassing months or years of development, but EUAs remain in effect only for the duration of the "emergency." It remains to be seen whether regulators will apply lessons learned during 2020 toward revising the risk-benefit algorithms they apply to future drug and device approvals, perhaps leading to more streamlined licensing of critical products under their purview.
PRNT and improved PRNT
Coronavirus NAbs are unique, among the various immunoglobulins raised in response to infection, in that they block the interaction between the coronavirus spike protein receptor binding domain and its cognate receptor, ACE2. Or, they prevent viruses from transferring genes to host cells. Either way, interpreting semi-quantitative NAb assays depends on where the designated signal cutoff is set.
"Assays like ELISA, lateral flow, and bead-based assays detect binding, not functional inhibition of infection," says Anis H. Khimani, Senior Strategy and Market Segment Leader at PerkinElmer. "An ideal assay measures NAb levels as well as their activity in blocking infection."
PerkinElmer's EUROIMMUN business unit recently released a test, Anti-SARS-CoV-2 QuantiVac ELISA (IgG), which incorporates a recombinant S1 subunit of the SARS-CoV-2 spike protein. "This assay detects and quantifies anti-S1 IgG antibodies in standardized, comparable units referenced to the World Health Organization's standard for Anti-SARS-CoV-2 IgG, and correlates well with a FDA EUA Neutralization Antibody Detection Kit," Fechner says. "This assay has already been CE-marked and we plan to file a request for its emergency use authorization with the FDA."
The gold standard for neutralization assays is the plaque reduction neutralization test (PRNT), which uses virus suspension and appropriate host cells to detect levels of infection or neutralization. PRNT is cumbersome and time-consuming, and requires Biosafety Level 3 facilities, which is why higher-throughput and more convenient ELISA-based NAb assay kits are more generally used.
PRNT assays have been developed with automated plate-imaging capabilities, which provide assay throughput while maintaining test accuracy and consistency. "Migrating assays from the conventional 24-well plate assay format to 96-well or higher formats, combined with imaging capabilities and algorithm-supported data analysis, significantly enhances the value of NAb detection," Khimani says. General accessibility and utility of PRNT assays is enhanced as well.
ACROBiosystems has launched a ready-to-use neutralizing antibody titer serologic assay kit that can sensitively and specifically detect COVID-19 neutralizing antibodies in 2 hours, showing a high correlation with FDA EUA approved ELISA kit.
Side-stepping
In addition to surveillance and therapeutic uses, NAb assays are used during clinical trials to quantify vaccine-induced or post-treatment protective immunity. "NAb assays can also assess the effectiveness of vaccines in generating immunity against novel strains," says Prajwal Paudel, Product Development Scientist at ACROBiosystems. “NAb tests cannot, however, diagnose active infection. ACROBiosystems has also received CE Mark for our assay kit that measures NAb blocking of SARS-CoV-2 RBD binding to ACE2 protein.
An EUA designation sidesteps the lengthy 510k process for approving IVDs but still sets criteria on sample qualification, collection and processing methods, assay performance and result interpretation, plus requires disclosures of test principle, assay steps, product manufacturing, as well as control(s) and calibration materials. "Particularly for COVID-19 serologic tests, regulators require evaluation in at least 30 unique SARS-CoV-2 RT-PCR positive patients as well as 75 negative samples to provide results with 95% confidence interval," Paudel tells Biocompare. Tests must demonstrate sensitivity with 90% positive percent agreement and 95% negative percent agreement with the comparator serum PRNT assay, and assay cutoffs should be established using independent samples.
Cross-reactivity with 12 viral infection samples that include influenza, common cold coronavirus, HIV, and respiratory syncytial virus is encouraged to be assessed as well, Paudel says, and recommend to include at least 10 anti-HIV positive samples. "Currently there is no requirement to provide mutant strain reactivity or inclusivity in the EUA application. Not yet, but how those variants affect the NAb protectivity in vaccinated and recovered population need to be closely monitored. ACROBiosystems have been quickly developing SARS-CoV-2 mutant proteins to support these kinds of studies, such as the UK B1.1.7 and South African variants."
(Source: https://www.biocompare.com/Editorial-Articles/572420-Neutralizing-Antibodies-in-the-Fight-against-COVID-19/)
 Popular ArticlesRelated RecommendationsPopular Events
Popular ArticlesRelated RecommendationsPopular Events
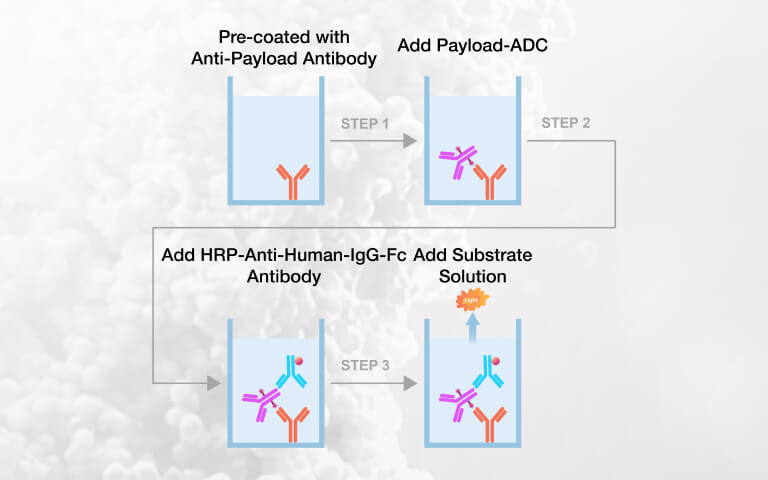 Payload-Specific ELISA Kits for Intact ADC Quantification in Preclinical PK Studies2026-07-22Page Views:49
Payload-Specific ELISA Kits for Intact ADC Quantification in Preclinical PK Studies2026-07-22Page Views:49 Expanding the Potential of Hematopoietic Stem Cells (HSCs): Emerging Strategies for HSCs Expansion and Clinical Manufacturing2026-07-22Page Views:33
Expanding the Potential of Hematopoietic Stem Cells (HSCs): Emerging Strategies for HSCs Expansion and Clinical Manufacturing2026-07-22Page Views:33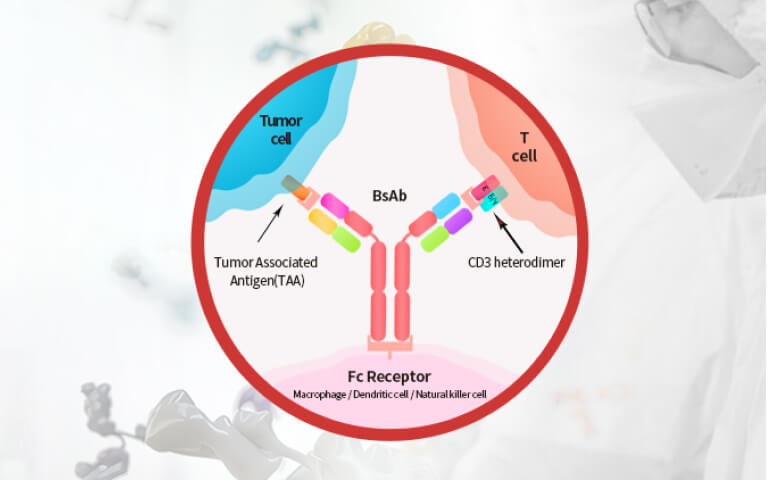 Advances and Challenges of BCMA×CD3 Bispecific Antibodies in Multiple Myeloma2026-07-21Page Views:101
Advances and Challenges of BCMA×CD3 Bispecific Antibodies in Multiple Myeloma2026-07-21Page Views:101 E. coli Host Cell DNA Testing: Independent Eurofins Validation Demonstrates Reliable Residual DNA Quantification2026-07-20Page Views:86
E. coli Host Cell DNA Testing: Independent Eurofins Validation Demonstrates Reliable Residual DNA Quantification2026-07-20Page Views:86 What Determines FcγR-CR T Function Beyond the Therapeutic Antibody?2026-07-14Page Views:61
What Determines FcγR-CR T Function Beyond the Therapeutic Antibody?2026-07-14Page Views:61




![[Free Sample] Universal Fluorescent Anti-VHH Antibody — Precise Detection of VHH-Based CAR Expression](https://console.acrobiosystems.com/uploads/demo/2026/06/02/Anti-VHHAntibodySmallbanner.jpg)
 Related Products
Related Products


