Leave message
Can’t find what you’re looking for?
Fill out this form to inquire about our custom protein services!
Inquire about our Custom Services >>






























 Order Online! Now! Get your $50 coupon for online order.
Order Online! Now! Get your $50 coupon for online order. Order Online! Now! Get your $50 coupon for online order.
Order Online! Now! Get your $50 coupon for online order.
Request a FREE sample of our GMP products! Request a FREE sample of our GMP products!
Fill out organ-on-a-chip questionnaire to win a FREE gift! Fill out organ-on-a-chip questionnaire to win a FREE gift!
> Key Raw Materials for Cell and Gene Therapy Finding the right supplier of raw materials can be a challenge. As you begin to consider pilot production and scale-up manufacturing, your supplier is not just a supplier but a partner in your manufacturing journey.
Qualifying GMP or cGMP (current Good Manufacturing Practice) raw materials is a responsibility that falls upon cell therapy manufacturers. Finding a good supplier that is able to support the transition to clinical manufacturing is crucial in regulated manufacturing. Our years of expertise in protein manufacturing, stringent quality control, and regulatory support allow us to offer industry-leading GMP proteins for ancillary use.

Raw or ancillary materials are materials used for the manufacturing of a pharmaceutical product. They are NOT intended to be a part of the final product formulation or as an Active Pharmaceutical Ingredient. As your cell therapy moves closer to the CMC stage, including pilot production, process optimization, and scale-up manufacturing, having a well-documented raw material is necessary to transition into clinical manufacturing. GMP-grade raw materials are designed to document the entire manufacturing facility, processes, and operators crucial for regulatory agencies to ensure it meets relevant guidelines.
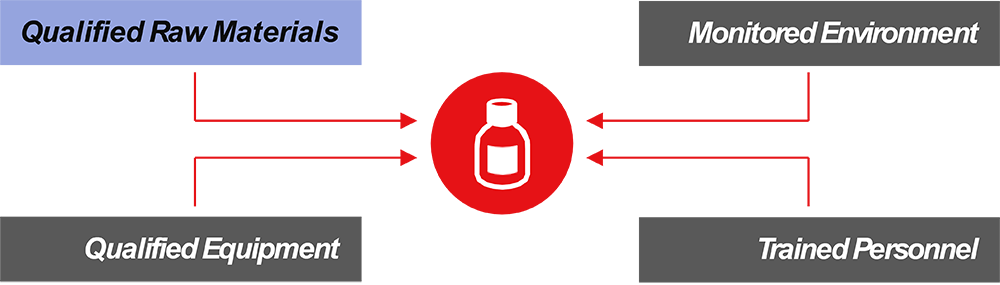
Qualified raw materials make all the difference. When selecting higher grade materials like GMP grade, we ensure that our raw material has undergone extensive QC testing, manufacturing process controls, and a complete set of documentation. This translates into tangible benefits for not only your therapy, but also your patients.


Incorporating GMP grade reagents in the preclinical stage or during pilot production avoids the need to re-validate and perform equivalency studies when moving into CMC manufacturing. These studies can be costly and time-consuming to perform and can easily outweigh the additional cost of incorporating GMP raw materials in the earlier development stages.
Learn More About GMP Grade Products
We designed our Premium (Pre-GMP) grade solutions as an intermediate grade for users that are looking to transition into clinical manufacturing but are not quite at that development stage. These solutions are manufactured using the same source, process, and quality controls, with the exception of the more costly GMP requirements. They perform the same as their GMP counterparts, helping users easily transition to GMP grade when they reach the clinical manufacturing stage.
Learn More About GMP Grade Products
Want to learn more about
your GMP Protein options?
Finding the right supplier is the same as finding a partner. Evaluating potential supplier’s capabilities to supporting your process is crucial to streamlining the transition from research-use only materials to GMP raw materials. With proper planning and clear, defined responsibilities, process optimization incorporating GMP reagents can be easily and quickly performed. Performing due diligence when evaluating a supplier means asking the right questions. Here’s some things to keep in mind when selecting a supplier:

Selecting a raw material supplier and implementing GMP-grade products earlier in the development process will prevent regulatory challenges when moving into clinical manufacturing. Talk to your supplier earlier about GMP to secure your raw materials and avoid future changes and consider finding and evaluating a secondary supplier to ensure your raw material supply.
Read our Article about Considerations in Selecting GMP-grade Raw Materials
Interested in testing
a GMP grade product?
Providing high-quality GMP products at an affordable cost can help lower the barrier in accessing innovative medicines including cell therapies. When transitioning from our premium (Pre-GMP) grade products to GMP, there should be minimal comparability testing needed. Our premium-grade products have been validated to have equivalent bioactivity and consistency and are manufactured using the same methods and processes.
Providing high-quality GMP products at an affordable cost can help lower the barrier in accessing innovative medicines including cell therapies. When transitioning from our premium (Pre-GMP) grade products to GMP, there should be minimal comparability testing needed. Our premium-grade products have been validated to have equivalent bioactivity and consistency, and are manufactured using the same methods and processes.
This includes products manufactured at our new facility. Our new facility was designed specifically for GMP production of proteins, enzymes, activation beads, and other solutions.



Equivalent bioactivity between Premium (Pre-GMP) vs GMP grade cytokines were measured using cell proliferation assays. (Left) Human IL-15 GMP (red) and premium (blue) saw no difference in CTLL-2 cell proliferation. (Right) Human IL-7 GMP(blue) and premium(red) saw no difference in proliferation of PBMC cells.
Human IL-2, GMP-grade
Human IL-7, GMP-grade
Human IL-15, GMP-grade
Explore >>
Premium (Pre-GMP) and GMP grade products are manufactured from the exact same source, process, and conditions. This means that the quality and performance are identical between both grades. The difference stems from a more comprehensive QC testing panel along with stringent specifications and documentation for our GMP products. For users earlier in the development stage, premium (pre-GMP) proteins are a cost-effective alternative that still ensures a smooth transition to clinical manufacturing.
| Premium Grade Products | GMP Grade Products | |
|---|---|---|
| Application | Early-stage R&D and preclinical studies are seamlessly linked to GMP level | Designed to meet clinical-stage needs for commercial manufacturing |
| Production Quality System | ISO9001/ISO13485 Quality certification system | ISO9001/ISO13485 quality certification system (R&D stage) + GMP quality system (production stage) |
| Production Quality System | ISO-certified general grade factory | GMP clean plant certified by the third-party authority |
| Stable cell lines | Validated cell lines thorough external inspection according to pharmacopoeia requirements to ensure no exogenous contaminations. | |
| Most AOF raw materials | AOF raw materials | |
| Pharmaceutical grade key excipients | Pharmaceutical grade key excipients | |
| Strict secondary sterilization filtrations | Strict secondary sterilization filtration | |
| C+A manual aseptic filling (ISO 5) | B+A fully automatic aseptic filling | |
| No specific virus removal/inactivation process | Specific virus removal/inactivation process steps (nanofiltration and low pH) | |
| Quality Control | Sterility/mycoplasma testing | Sterility/mycoplasma testing |
| Endotoxin control and testing | Endotoxin control and testing | |
| Key production equipment/analytical instruments are validated | Production equipment/analytical instruments/key analytical methods are rigorously validated, and analytical instruments are audited. | |
| Process-related impurities: DNA, HCP, process residues | Process-related impurities: DNA, HCP, process residues | |
| No additional safety tests | Increased related virus residues, animal safety experiments in vivo (abnormal toxicity, acute toxicity, etc.) | |
| Supporting Documentation | Necessary documents or declarations | Comprehensive Secondary Regulatory Support Documentation |
| No DMF filing | DMF filing for all products |
Premium (Pre-GMP) and GMP grade products are manufactured from the exact same source, process, and conditions. This means that the quality and performance are identical between both grades. The difference stems from a more comprehensive QC testing panel along with stringent specifications and documentation for our GMP products. For users earlier in the development stage, premium (pre-GMP) proteins are a cost-effective alternative that still ensures a smooth transition to clinical manufacturing.
Sometimes it can be a challenge to find the right raw materials that you need, especially with innovative therapies. Instead of trying to produce it in-house, rely on our experienced custom GMP service project team! Our team here at ACROBiosystems will assign a dedicated project manager to keep track of your needs and ensure delivery on time. We offer different GMP grade services to assist you both in the short-term and in the future. This includes:
When it comes to preparing the transition from RUO to GMP grade proteins, it can be hard to know exactly when to start switching. We answer a few of the more common questions that we get in the industry to help shed some insight about the regulatory landscape of utilizing GMP raw materials and enter the clinical phase without any worries.
Raw materials should be sourced from qualified suppliers following a pre-established supplier qualification and monitoring process. We perform annual risk assessments for raw materials used in our GMP process along with our suppliers. This encompasses visual inspections and documentation tracking of associated documents including certificates of analysis (CoA), Certificates of Origin (CoO), TSE/BSE declarations, and animal-free statements, when applicable. Before acceptance, key raw materials are always identified and validated.
We advise cell therapy manufacturers to source either pharmaceutical or GMP-grade whenever possible. Our GMP-grade products are manufactured and tested in accordance with relevant international standards. * Raw material suppliers should have a certified quality management system (QMS) along with an ISO 9001 / 13485 certification. Independent audits, quality policies, and quality processes should be established to ensure quality that matches customer expectations. Cell therapy manufacturers should also perform due diligence by qualifying their suppliers through questionnaires, on-time delivery, SCAR, and other auditing activities. Documentation including certificates of analysis, certificates of origin, and other audit trail documentation should be received from each supplier and kept as records.
Cell therapy manufacturers should begin discussions with potential suppliers as soon as possible to avoid issues when entering production. Ensuring raw material suppliers have the ability to scale-up can prevent any last-minute changes that could require expensive revalidation studies.
Identifying a secondary supplier is also crucial. Although raw materials may look identical, there is no guarantee on how it behaves in your biological system. Do not assume that switching suppliers can be easily done – subsequent validation studies showing equivalence between raw materials from different suppliers is required. Identifying a secondary supplier can be an insurance policy if any issues arise with your primary supplier.
The quality of manufactured cell and gene therapies is directly influenced by the raw materials used in their production, which, in turn, impacts the therapy’s impact and efficacy. Relevant regulations stipulate that the selection of raw materials should consider their necessity, rationality, and safety, preferably opting for materials approved for human use or meeting pharmacopoeia standards. Although safety and quality requirements for raw materials have lower priority in the initial preclinical stage, transitioning to regulatory-compliant GMP-grade materials becomes imperative during CMC or clinical conversion. This transition involves thorough testing, safety assessments, and process validation, demanding considerable time and effort.
If the same supplier is utilized, the physical properties and biological activity of premium-grade (PG) raw material ideally mirror those of GMP-grade, differing mainly in regulatory support documentation. Rigorous safety testing of materials interacting with patients is crucial, and confirming the consistency of manufacturing processes between PG and GMP materials is recommended. We ensure that both our PG and GMP products are derived from the same cell bank source, undergo strict quality control, demonstrating biological identity through extensive trials.
For clinical applications, an early shift to GMP is recommended, even during the preclinical phase, facilitating equivalence testing at a more manageable stage. Opting for an experienced supplier well-versed in quality management, protein biochemistry, analytical testing, and regulatory affairs is advantageous. Experienced suppliers aid in navigating changing regulatory environments, providing necessary documentation like a Drug Master File (DMF) to streamline regulatory processes. Audits of supplier facilities, preferably in person or online, strengthen the strategic partnership between suppliers and customers. Maintaining strict adherence to approved operating procedures, at least for three consecutive batches, ensures consistency and compliance with predetermined acceptance criteria for our PG products developed through the GMP product line.
Biological reagents are notorious when it comes to susceptibility to variability, especially cytokines and growth factors. Only after these reagents have been successfully developed and validated through multiple successive batches meeting the stringent outlined specifications, along with the establishment of a stable process and rigorous conformance assessment, can the product be moved into large-scale manufacturing and commercialized. This process forms a robust foundation in ensuring the consistency and reliability of GMP raw materials.
Furthermore, the manufacturing of products occurs within GMP facilities and under GMP quality systems, with strict control implemented through documented Standard Operating Procedures (SOPs) and trained personnel. This crucial information should be disclosed during audits. It is reasonable to request data from multiple batches to evaluate a supplier's capability to consistently produce a reproducible protein. Ideally, materials should be sampled from various batches to assess their consistency across the entire system. We identify standard batches and compare each new production batch to the established standard before releasing it to the market. This approach mitigates variability and ensures consistent product performance. Currently, we maintain excellent control over lot-to-lot differences, specifically in biological activity, within the range of 10-15%.
Safety regarding any raw materials employed in cell and gene therapy manufacturing is critical to ensure final therapeutic quality. This emphasizes the need for safety control to prevent contamination by exogenous factors. These ‘exogenous’ factors refer to inoculants, cell matrices, residual raw materials during production, bacteria, fungi, mycoplasma, and viruses.
The FDA recommendation of using the highest-quality materials means that all raw material suppliers should ensure that their materials or reagents are produced under the appropriate quality standards to mitigate any unreasonable risks to patients. This means that strict quality control of raw materials used in CGT production is a necessary measure to reduce the risk of external contamination and ensure the safety and efficacy of cell and gene therapy products. Expanding upon this requirement, the following requirements are outlined in various international pharmacopeias, including:
Our resident team of GMP professionals has decades of experience combined interpreting and implementing regulatory guidelines. This is essential for novel therapeutics where responsibilities between raw material suppliers and cell therapy manufacturers may not be clear, or a distinct set of validation studies is required to remain compliant.
Our products are designed to follow global quality regulations from the NMPA, FDA, EMA, PMDA, and other regulatory agencies. Each facility is also ISO-certified and GMP-compliant. When it comes to documentation, we provide two levels of documentation based on each customer’s need.
Overall, our GMP product lines encompass FDA records support such as drug-master files. If there are any issues with obtaining and IND or BLA, please contact quality@acrobiosystems.com to obtain a letter of authorization (LOA). Any other support can be done online or on-site, free of charge, including any responses to regulatory questions related to raw materials.
Learn More About our GMP Capabilities, Quality, and Regulatory Support
Making the Transition to GMP Brochure
GMP Grade Solutions for Cell Manufacturing
Custom GMP Protein Services
Special Topic on Deep Interpretation of GMP Product Quality 1
Speak with our Quality Engineers



This web search service is supported by Google Inc.
















 A-Z
A-Z